1. Apparato respiratorio
Boccheggiante
|
ga
- gasper |
Legato
al sesso e recessivo
Gruppo di associazione V - cromosoma Z
In letteratura è riportata una
sola mutazione che colpisce il sistema respiratorio. Price (1966)
ha
fornito una breve descrizione di un’anomalia respiratoria: dopo poco tempo
dalla nascita i pulcini presentavano un rantolo bronchiale e un caratteristico
boccheggiamento che entro 4 giorni diventavano imponenti, seguiti da un’elevata
mortalità. I soggetti che riuscirono a diventare adulti continuarono a
presentare un rantolo bronchiale e non raggiunsero un peso normale.
Non fu possibile isolare alcun agente infettivo e non fu
riscontrato alcun segno di processo infiammatorio.
2. Apparato riproduttivo
2.1. Ipoplasia gonadica ereditaria
|
Hgh
-
hereditary
gonadal hypoplasia |
Autosomico
incompletamente dominante
Gruppo di associazione sconosciuto
Questa anomalia descritta da
Lojda e Hovorka (1968) colpisce solo i maschi: bassa concentrazione di
spermatozoi con una relativamente alta percentuale di forme patologiche nel
seme di un maschio di Livorno bianca che presentava un’ipoplasia
testicolare. La fertilità di questo maschio era significativamente inferiore
a quella dei maschi normali suoi consanguinei.
2.2. Atresia degli organi riproduttivi
|
Aro
-
atresia
of reproductive organs |
Autosomico
dominante
Gruppo di associazione sconosciuto
Una chiusura ereditaria dell’ovidutto
in Livorno bianca fu descritta da Finne e Vike (1951):
queste galline non erano in grado di deporre uova a causa di
una rottura dell’ovidutto a livello dell’istmo, spesso accompagnata da un
restringimento fra vagina e cloaca. L’ovaio era sviluppato in modo
normale e l’istinto di deposizione era molto evidente, ma appena dopo la maturità sessuale queste femmine mostravano segni di
ovodeposizione interna con susseguente elevata mortalità.
All’esame necroscopico del padre si
mise in evidenza un deferente di destra discontinuo, associato a un testicolo
destro floscio e raggrinzito. Quindi questa mutazione colpisce sia i maschi
che le femmine.
Furono prodotte altre generazioni di femmine affette da
quest'anomalia, potendosi altresì dimostrare che la condizione era
determinata da un singolo gene dominante, ma dal momento che le femmine
affette dalla patologia non erano in grado di deporre uova, non fu mai
possibile dimostrare se il gene in causa era autosomico oppure legato al
sesso, né fu possibile ottenere maschi omozigoti per studi ulteriori.
2.3. Sviluppo dell’ovidutto destro
|
Rov
-
right
oviduct development |
Autosomico
incompletamente dominante
Gruppo di associazione sconosciuto
In base alle revisioni della
letteratura a opera di Sell (1959) e di McBride (1962)
sono
stati descritti numerosi casi di galline dotate di due ovidutti. In condizioni
normali solo l’ovidutto di sinistra è ritenuto un organo funzionalmente
attivo, essendo quello destro un residuo privo di funzione attaccato alla
cloaca e che solo in condizioni anormali aumenta di dimensioni e si riempie di
fluidi.
McBride ha suddiviso in 4 classi i casi riportati in
letteratura:
1 - ovaio e ovidutto sia destro
che sinistro completamente sviluppati
2 - ovidutto destro sviluppato
con un accenno di sviluppo dell’ovaio destro
3 - ovidutto sia destro che
sinistro sviluppati, ma con assenza completa dell’ovaio destro
4 - ovaio e ovidutto di sinistra
normali con un ovidutto destro imperfetto, abitualmente cistico.
Le ultime due classi includono gran parte dei casi
riportati in letteratura. È significativo il fatto che in letteratura non è
descritta alcuna femmina dotata di un sistema riproduttivo solo dal lato
destro.
Ricorrendo agli opportuni test si è potuto dimostrare trattarsi di un’anomalia dovuta a un singolo gene autosomico incompletamente dominante.
Quasi tutte
le femmine di uccello hanno solo l'ovaio sinistro funzionante. Un'eccezione è
costituita dallo Sparviero![]() ,
Accipiter
nisus, nonché da altri Falconiformi
,
Accipiter
nisus, nonché da altri Falconiformi![]() , cui dobbiamo aggiungere il Kiwi
, cui dobbiamo aggiungere il Kiwi![]() , che appartiene al genere Apteryx.
Ma solo l'ovidutto sinistro è funzionante, essendo quello destro per lo più
inutilizzato, e talora vestigiale. Si veda
per completezza - nella pagina dello Sparviero, del Falco e del Kiwi - la
ricerca di Kinsky (1971) The consistent presence of paired ovaries in the Kiwi (Apteryx)
with some discussion of this condition in other birds.
, che appartiene al genere Apteryx.
Ma solo l'ovidutto sinistro è funzionante, essendo quello destro per lo più
inutilizzato, e talora vestigiale. Si veda
per completezza - nella pagina dello Sparviero, del Falco e del Kiwi - la
ricerca di Kinsky (1971) The consistent presence of paired ovaries in the Kiwi (Apteryx)
with some discussion of this condition in other birds.
2.4. Ridotta ovulazione
|
ro
- restricted
ovulator |
Legato
al sesso, recessivo
Gruppo di associazione V - cromosoma Z
Questa mutazione è già stata
descritta in questo volume: VIII.8.5.
2.5. Carenza di riboflavina
|
rd
-
riboflavin
deficiency |
Autosomico,
recessivo
®
incompletamente dominante
Gruppo di associazione sconosciuto
Notizie dettagliate relative a
questa mutazione si trovano nel II volume - XVII.8.2.
e in questo volume: VIII.8.13.
3. Apparato urinario
3.1. Ipoplasia renale
|
kh
-
kidney
hypoplasia |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
L’ipoplasia renale nel pollo
è stata studiata da due gruppi di ricercatori. Jeffrey (1937)
trovò
in un ceppo di Livorno bianca l’assenza o l’atrofia del rene sinistro
senza conseguenze apparenti per lo stato di benessere. Non è stato possibile
determinare con sicurezza che si trattasse di un’anomalia su base genetica.
Pun (1961) studiò una situazione di ipoplasia renale nella Livorno
perniciata che pareva dovuta a un singolo gene recessivo dotato di penetranza
ed espressività variabili, con un’espressività maggiore nelle femmine
rispetto ai maschi. L’ipoplasia non prediligeva il rene destro più del
sinistro e nel rene normale era presente un’ipertrofia compensatoria.
3.2. Gotta ereditaria
|
go
-
gout |
Autosomico,
recessivo
Probabile gruppo di associazione II - cromosoma 2
La gotta può occasionalmente
manifestarsi negli uccelli in caso di dieta particolarmente ricca in proteine,
con sviluppo di tofi gottosi a livello delle articolazioni dei piedi e delle ginocchia, associati a elevati tassi plasmatici di acido urico.
Nel pollo è stato possibile dimostrare che pur con una dieta a basso tenore
proteico può verificarsi uno stato gottoso associato a iperuricemia. In base
alle osservazioni effettuate si ipotizza che questo gene abbia un rapporto di
linkage con il locus i+.
4. Sistema ormonale
4.1. Nanismo tiroideo
|
td
-
thyrogenous
dwarfism |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
Questa particolare forma di
nanismo fu riportata per la prima volta da Landauer (1929)
in
un solo soggetto di Rhode Island Red, il quale si presentava di dimensioni
ridotte e il cui piumaggio era più lungo del normale. La tiroide era
ingrandita ed era formata da tessuto aplastico privo di colloide. Tale
situazione di ipotiroidismo ricordava parecchio la condizione umana nota come
mixedema infantile.
Anche Mayhew e Upp (1932) comunicarono
una condizione di nanismo su base ipotiroidea nella RIR, la cui descrizione
somigliava a quella fornita da Landauer. Alla schiusa i pulcini apparivano
normali, ma delle differenze sostanziali cominciarono a manifestarsi a 3
settimane di vita: le zampe erano molto corte in rapporto alle dimensioni del
corpo e le dita più esterne erano girate all’infuori e all’indietro, la
testa era più slargata in corrispondenza delle regione oculare e il becco era
simile a quello di un pappagallo. Inoltre la coda era portata a un livello
corrispondente all’altezza della parte centrale del corpo, con piume dirette
in basso. I pulcini erano socievoli e attivi, ma difficili da allevare fino
alla maturità, e nessun soggetto nano la raggiunse.
Gli studiosi furono in grado di concludere per una
patologia causata da un gene autosomico recessivo allo stato omozigote, il cui
simbolo td fu proposto da Hutt (1949).
4.2. Tiroidite autoimmune
|
Tiroidite
autoimmune ereditaria |
Poligenico
Il ceppo obeso di Livorno bianca
- OS, obese strain - descritto da Cole (1966) fu
sviluppato presso la Cornell University da 20 femmine obese che erano
segregate dal Cornell C strain - CS - durante la stagione di schiusa 1955-1957.
Attraverso la selezione l’incidenza di questa obesità venne incrementata
dallo 0% a oltre l’80% nei maschi e da meno dell’1% a oltre il 90% nelle
femmine. L’espressione dell’obesità era piuttosto più frequente e più
pronunciata nelle femmine rispetto ai maschi. L’obesità è stata la prima
caratteristica a essere osservata in questi soggetti, da cui deriva il nome
assegnato al ceppo selezionato.
I soggetti obesi venivano generalmente riconosciuti come
tali a partire da 6-10 settimane di vita e avevano le caratteristiche che
tipicamente si associano a uno stato di ipotiroidismo: dimensioni ridotte,
pelle soffice e pienotta sollevabile in pliche, abbondante accumulo di adipe,
struttura setosa del piumaggio che si presentava eccessivamente lungo. Il
comportamento era tranquillo ed era presente una particolare sensibilità alle
basse temperature. La maturità sessuale si dimostrò ritardata e la
deposizione di uova fu scarsa, con normale volume delle uova. Fertilità e
schiudibilità generalmente normali.
Fu Cole (1968) a dimostrare per primo l’origine
autoimmune di questa condizione patologica, trattandosi di una tiroidite
autoimmune spontanea, ampiamente impiegata come modello per la tiroidite di
Hashimoto in campo umano, che è una tiroidite cronica a patogenesi
autoimmune.
Cole giunse alla conclusione che questa tiroidite dei
polli è ereditata come tratto poligenico, che non è completamente recessivo.
Secondo Hala (1988) potrebbe trattarsi della messa in gioco di 5 geni
maggiori nel regolare l’espressione completa della tiroidite: due famiglie
di geni in cui due geni di una famiglia codificano per un’anormale
reattività del sistema immunogeno, e tre geni della seconda famiglia, uno dei
quali recessivo, codificano per la suscettibilità dell’organo bersaglio nei
confronti dell’attacco autoimmune. Sarebbero implicati anche geni
modificatori minori.
Dell’OS abbiamo già succintamente parlato nel vol.II
- XIX.9.3. quando abbiamo analizzato i linfociti TS,
cioè i linfociti T soppressori: queste cellule sono particolarmente
importanti nel sopprimere la risposta agli autoantigeni, assicurando una
risposta immunitaria anticorpale solo verso antigeni esterni, per cui una
carenza o una mancanza di linfociti TS
comporta malattie autoimmuni nelle quali l’organismo arriva a distruggere
parte dei propri tessuti; situazione che si verifica sia nell’uomo che nel
pollo, come appunto nell’OS che presenta una tiroidite autoimmune.
4.3. Tiroidite indotta dalla dieta
|
Tiroidite
indotta dalla dieta |
Poligenico
Il ceppo C della Cornell - CS -
mostrava un tasso sorprendentemente elevato di autoanticorpi anti ormone
tiroideo - T3 e T4 - e anti tireoglobulina.
L’aggiunta di iodio alla dieta durante le prime 10 settimane di vita fu in
grado di incrementare notevolmente l’incidenza di tiroidite autoimmune in
questi soggetti, come fu possibile dimostrare dall’esame istologico della
tiroide e dal tasso di autoanticorpi, mentre una dieta carente in iodio aveva
come risultato una riduzione dell’incidenza della malattia. Si è potuto
dimostrare che gli elevati livelli dietetici di iodio sono in grado di
incrementare l’immunogeneticità della molecola di tireoglobulina.
Questa particolare suscettibilità del CS alla tiroidite
autoimmune indotta dallo iodio è senza dubbio su base genetica e
probabilmente riconosce un meccanismo multifattoriale. La condizione del CS è
diversa da quella descritta nell’OS, nel quale la produzione di
autoanticorpi è indipendente dal tasso di iodio presente nella dieta.
4.4. Diabete insipido
|
di
-
diabetes
insipidus |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
Buss e Murphy (1965)
riferirono
di un ceppo di Livorno bianca caratterizzato da polidipsia e poliuria, cioè
da esagerata sete ed elevata produzione di urina; l’esame della genealogia
suggeriva una causa verosimilmente genetica e gli incroci successivamente
eseguiti dimostrarono trattarsi di un singolo gene autosomico recessivo.
I soggetti omozigoti di/di apparivano
normali quanto a vitalità, fertilità, consumo di cibo, peso corporeo, numero
e peso delle uova, spessore del guscio e qualità dell’albume. Essi tuttavia
mostravano un grado elevato e variabile di polidipsia e poliuria, che a 3
settimane di vita comportava un’assunzione di liquidi circa tre volte
superiore al normale; la pressione osmotica del plasma e la concentrazione
plasmatica di sodio erano tuttavia molto simili nei soggetti normali e in
quelli con polidipsia.
Sull’effettivo meccanismo di questo diabete insipido non
è stato raggiunto un accordo unanime. Infatti, se una serie di indagini è
stata in grado di dimostrare una sensibilità alla somministrazione di agenti
antidiuretici - come vasopressina e vasotocina -, altri studiosi hanno
concluso trattarsi di una forma di diabete insipido in cui è in causa un
semplice malfunzionamento renale senza implicazioni ormonali.
5. Apparato visivo
Anatomia e fisiologia del globo
oculare sono state descritte in vol.II - XXVI.5.
5.1. Microftalmia bilaterale
|
mi,
Mi-2, mi-3 -
bilateral
microphthalmia |
Autosomici
Gruppo di associazione sconosciuto
Sono stati descritti almeno 3
casi di microftalmia, cioè di una condizione patologica caratterizzata da
rimpicciolimento del globo oculare. Jeffrey (1941) fu
il primo a riferire una microftalmia bilaterale nella Plymouth Rock barrata,
ereditata come carattere autosomico recessivo. I soggetti che ne erano affetti
presentavano un globo oculare il cui diametro era circa la metà del normale e
che non protrudeva all’esterno, accompagnato da una marcata depressione
della regione oculare su ogni lato della testa. Si accompagnava anche una
riduzione del volume della cresta, che era assottigliata e talora sdoppiata.
La schiusa degli embrioni che ne erano affetti si aggirava sul 25-34% e
nessuno dei pulcini raggiunse la maturità. Si ritenne che la mortalità dei
pulcini fosse dovuta all’impossibilità di trovare acqua e cibo a causa
della cecità, ma molto verosimilmente la mortalità di questi soggetti
microftalmici era dovuta a un effetto pleiotropico. Somes (1980) assegnò il simbolo mi a
questa mutazione.
Wight e Carr (1965) osservarono
una Livorno perniciata normale produrre una progenie affetta da microftalmia
mono o bilaterale in due periodi differenti quando venne inseminata
artificialmente con seme di maschi differenti e senza rapporto di parentela.
Dei 59 pulcini che nacquero, 18 - e quindi il 33% - erano anormali: il difetto
variava da una anoftalmia monolaterale - assenza di qualsiasi struttura
oculare da un solo lato - a occhi di dimensioni appena più piccoli della
norma. Nessuno dei pulcini affetti sopravvisse per potersi riprodurre; tre
femmine e un maschio normali nati da questa femmina ebbero una progenie ma non
ne nacquero pulcini anormali. Gli autori conclusero per un tratto dominante
con penetranza incompleta - Mi-2 -, ma la prova addotta non era conclusiva.
Non bisogna dimenticare, come puntualizzò a suo tempo Jeffrey, che parecchi
casi di microftalmia unilaterale nel pollo e in altri animali non sono di
origine genetica.
Finzi e Romboli (1978) riscontrarono
un’altra microftalmia recessiva - mi-3 - nella New Hampshire, che sembra
essere diversa da quella descritta da Jeffrey. Anche se gli autori non l’hanno
specificato, è implicito che la microftalmia era bilaterale. Come per il gene
mi, il gene mi-3 comportò una mortalità del 30-45% prima della
schiusa, ma permise ai soggetti di diventare adulti e di riprodursi
artificialmente. Inoltre, gli occhi di alcuni soggetti mi-3 si avvicinavano
alla norma, mentre in altri erano drasticamente ridotti di volume. La cresta
non veniva alterata, ma in compenso si notò che le piume del collo erano
rigirate lungo il rachide, dando l’impressione di un ciuffo, che nei maschi
si presentava dietro la testa e nelle femmine ai lati del collo. Questa
situazione potrebbe essere un effetto pleiotropico di mi-3.
5.2. Occhio spalancato
|
pop
-
pop
eye |
Legato
al sesso, recessivo
Gruppo di associazione V - cromosoma Z
Le caratteristiche fenotipiche
di questa mutazione legata al sesso consistono in una protrusione bilaterale
della cornea di entità da lieve a severa e che può essere rilevata a partire
da 5 settimane di vita.
Nel soggetto adulto la spiccata protrusione della cornea
ha come conseguenza un aumento della profondità della camera anteriore dell’occhio
e cicatrici corneali al di sotto dell’epitelio. Queste alterazioni non
sfociano in cecità, ma sembrano essere responsabili di un astigmatismo. Nell’uomo
questa situazione patologica è nota come cheratocono, cioè cornea foggiata a
cono.
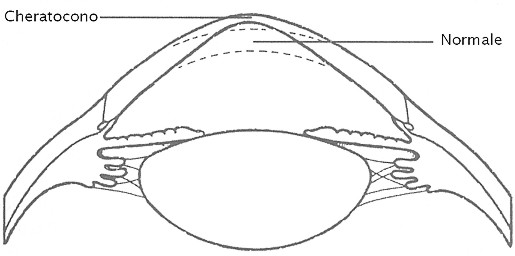
Fig. XVII. 1 - Curvatura e spessore della cornea normale e in caso di cheratocono
Lo studio istologico della mutazione pop ha
rivelato una graduale degenerazione delle cellule basali dell’epitelio
corneale; nei soggetti adulti l’epitelio corneale è ridotto a 2-3 strati di
cellule basali appiattite che giacciono su una membrana basale anormale; anche
le cellule dell’endotelio corneale sono differenti rispetto alla norma e la
perdita di cellule in relazione all’avanzare dell’età è più rapida
rispetto a quanto accade in condizioni normali.
Queste modificazioni distrofiche su base non infiammatoria
a carico della curvatura della cornea dovute ad assottigliamento della
porzione centrale e alla protrusione conica sono note appunto come
cheratocono, che ha caratteristiche molto simili al cheratocono umano. Dal
momento che la mutazione pop è l’unica causa nota di un cheratocono
in animali domestici, il suo studio potrebbe essere di notevole aiuto al fine
di comprendere l’eziologia e la patogenesi del cheratocono umano.
5.3. Globo oculare ingrandito con cecità
|
beg
-
blindness
enlarged globe |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
Il fenotipo di questi pulcini
consiste in cecità alla schiusa, accompagnata da aumentate dimensioni dei
globi oculari che danno luogo a vari gradi di esoftalmo.
Negli embrioni, a partire dall’8° giorno d’incubazione,
è possibile osservare istologicamente la progressiva comparsa di piccole
discontinuità a carico della retina le quali vanno progressivamente
aumentando in numero e dimensioni col progredire dell’incubazione. All’atto
della schiusa sono presenti numerosi spazi intercellulari a carico della
retina e la degenerazione retinica continua a progredire con il crescere del
pulcino, con interessamento dei fotorecettori e delle cellule pigmentate.
5.4. Displasia e degenerazione retinica parziali
|
rdd
-
retinal
dysplasia and degeneration |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
Questa mutazione venne descritta
per la prima volta da Randall e McLachlan (1979):
alla schiusa i pulcini presentavano una visione limitata e un’attività
inferiore alla norma. A 6 mesi d’età la maggior parte dei soggetti non
rispondeva agli stimoli visivi. Gli elettroretinogrammi in pulcini di una
settimana di vita indicavano che la capacità visiva era scarsa ma non ancora
persa, e che la sensibilità dei coni e dei bastoncelli era molto ridotta.
Le sezioni istologiche condotte in embrioni e in pulcini
appena nati hanno mostrato anomalie della retina che si estendono dall’epitelio
pigmentato alla lamina interna. A partire dall’8° giorno d'incubazione
nell’epitelio pigmentato furono osservate delle discontinuità che andarono
aumentando in numero e in dimensioni col procedere dell’incubazione, ma a
differenza delle discontinuità osservate nella mutazione beg esse
scomparvero nel giro di una settimana dalla nascita. Dopo la schiusa fu
osservata una progressiva degenerazione dei fotorecettori.
Questa mutazione può essere utile come modello nello
studio di anomalie umane dell’occhio, tipo la retinite pigmentosa. Ma
necessitano ulteriori dati circa la retina dei soggetti rdd.
5.4. Cecità coni e bastoncelli
|
rc
-
rods
and cones |
Autosomico,
recessivo
Gruppo di associazione sconosciuto
A riferire di questa mutazione
fu Cheng (1978) che per primo la osservò in una linea di polli
portatori di una traslocazione a carico del cromosoma Z e del cromosoma 3
indotta da etilmetansulfonato.
Questi pulcini erano ciechi alla nascita. Nonostante
fossero nati in modo normale, essi non rispondevano agli stimoli visivi e
mostravano casualmente inchini e torsioni della testa allo scopo di mantenersi
in equilibrio.
Pang (1988) misurò la concentrazione di
melatonina (vol.II - XXVIII.15.3.) nella
retina, nel siero e nella ghiandola pineale di pulcini con età compresa fra
le 4 e le 14 settimane di vita, usando come controllo pulcini eterozigoti di
età corrispondente e dotati di normale capacità visiva. Sia i mutanti che i
controlli mostravano una variazione diurna dei livelli di melatonina retinica
a tutte le età, con livelli notturni elevati. I pulcini mutanti presentavano
livelli di melatonina retinica al buio significativamente più bassi (38-75%) rispetto ai controlli della
stessa età. Questa riduzione della melatonina retinica sembra possa essere
correlata con la degenerazione dei fotorecettori retinici. Invece non si
misero in evidenza differenze significative tra i due gruppi riguardo ai tassi
di melatonina epifisaria. Tuttavia, quando si prese in esame la melatonina
serica dei due gruppi in studio, i pulcini mutanti avevano livelli di
melatonina circolante significativamente più bassi.
In conclusione: il gene rc agisce sui fotorecettori
e sulla melatonina retinici e possiede anche effetti pleiotropici sulla
melatonina circolante. Questi polli sono utili nello studio circa la sintesi,
la regolazione e le funzioni della melatonina sia come neuromodulatore oculare
sia come ormone con effetti sistemici, nonché in ricerche relative all’eziologia
e alla patogenesi della degenerazione retinica.
5.5. Amelanosi oculare e degenerazione retinica
|
Amelanosi
oculare e degenerazione retinica |
Poligenico
autosomico
Probabile associazione con il locus E
Il gruppo di soggetti affetti da
amelanosi ritardata - ceppo DAM = delayed amelanosis - fu ottenuta nel 1971 da
una femmina del ceppo sperimentale perniciato dell’Università del
Massachusetts. Questa femmina particolare possedeva un piumaggio giovanile
normalmente pigmentato, ma il piumaggio adulto divenne completamente bianco e
il soggetto andò incontro a cecità.
In seno al ceppo DAM l’incidenza dell’amelanosi
ritardata fu pari al 60%, con un 25% di soggetti anormali che presentava anche
cecità. Selezionando i soggetti ciechi, la frequenza della cecità salì all’81%.
La segregazione genetica indicava che la cecità era strettamente associata
all’amelanosi ritardata e che ambedue i tratti erano dovuti a geni
autosomici; si giunse a concludere per tratti poligenici in cui erano
implicati pochi geni che avevano una relazione con il locus E.
Ulteriori studi sull’eziologia di queste situazioni
patologiche misero in evidenza che i soggetti DAM possedevano un sistema
immunitario iperattivo, particolarmente a carico dei linfociti B che sono
responsabili della risposta immunitaria su base umorale attraverso la
produzione di tutte le immunoglobuline
solubili;
possedevano inoltre dei melanociti morfologicamente anormali. Dopo la nascita
i melanoblasti si differenziavano a ogni muta in melanociti progressivamente
più anormali che venivano eliminati dalle piume in rigenerazione. Inoltre la
coroide dell’occhio andava incontro a una perdita di pigmento melanico
attraverso un meccanismo autoimmune con massiva infiltrazione di leucociti
mononucleati in seno al connettivo. L’amelanosi oculare colpiva l’epitelio
pigmentato della retina portando a degenerazione retinica e cecità.
Il ceppo DAM sembra essere un prezioso modello per lo studio della vitiligine e delle malattie oculari umane che hanno relazione con la melanina.
|
sommario |
top |
avanti |
|
|
|